

What is Emergency Use Authorization (EUA)?
The most important Covid question no one understands. Let's fix that right now.

During the Covid pandemic, the U.S. government spent billions of dollars on nearly 400 products intended to protect, diagnose and treat hundreds of millions of people – all with the label “EUA” or “Emergency Use Authorization.”
But what does EUA actually mean?
Even before we answer that question, and by way of understanding where EUA stands in relation to other pathways for authorizing or approving medical products, it is helpful to look at what EUA is not:
EUA is not a designation for an experimental product undergoing a clinical trial
If we only understand one thing about EUA it should be this: EUA does not apply to a product undergoing a clinical trial governed by FDA regulations or other legal requirements.
EUA is also not the same as Expanded Access Use (EAU), often called “compassionate use” access, which applies to granting patients with severe, incurable diseases access to experimental products before they are fully approved.
This table from an FDA-CDC 2020 presentation summarizes the differences between products undergoing clinical trials, products given to patients through expanded “compassionate” access, and products authorized through EUA:
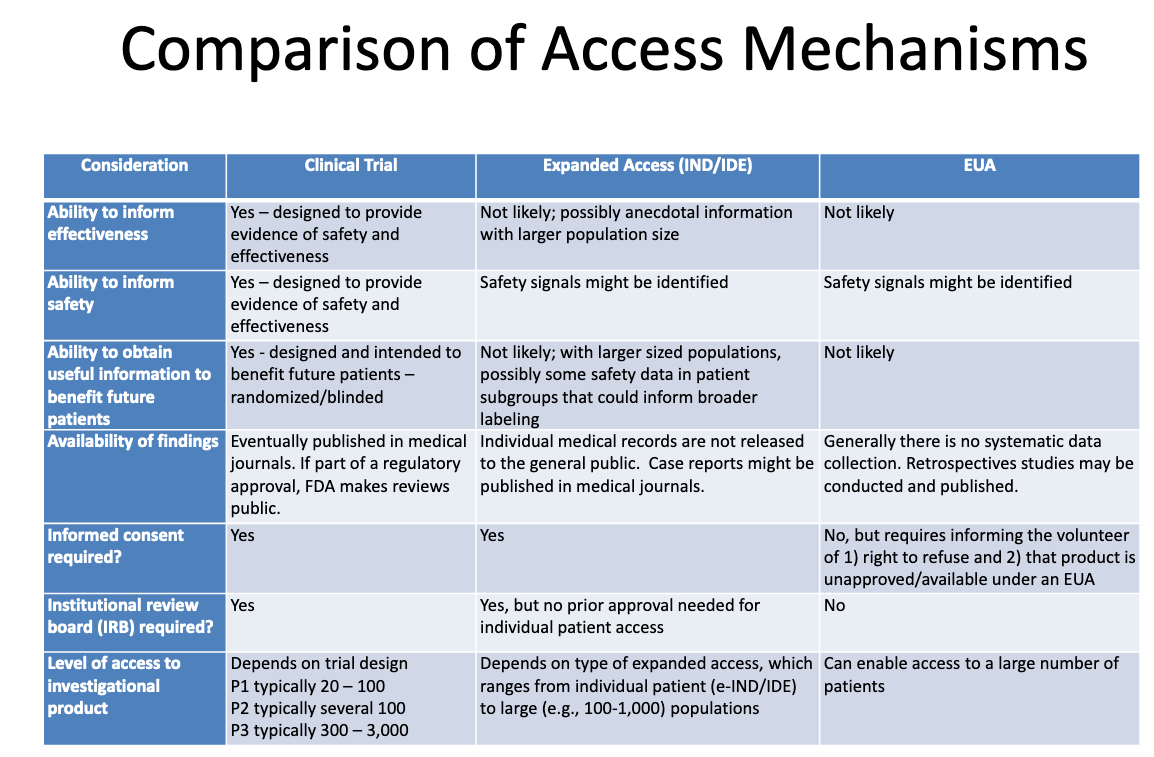
Here’s what this table tells us about EUA:
The process of granting EUA is not likely to generate any information about a product’s effectiveness.
The process of granting EUA is not designed to provide evidence of safety or effectiveness, but safety signals might be identified.
It is unlikely, once a product is granted EUA and administered to some patients, that any useful information will be obtained to benefit any future patients.
There is no systematic data collection on effectiveness or safety with EUA, and no data is published in medical journals as part of the regulatory approval process.
No informed consent is required, but patients who “volunteer” to take the product must be told they can refuse and that the product is unapproved/available under EUA.
No institutional review board (IRB) is required. [IRB is a board that is supposed to protect the well being of human subjects in clinical trials]
To clarify even further how separate EUA is from any normal approval process, in a 2009 Institute of Medicine of the National Academies publication, we find this statement:
It is important to recognize that an EUA is not part of the development pathway; it is an entirely separate entity that is used only during emergency situations and is not part of the drug approval process. (p. 28)
To summarize:
The process of granting a product EUA is unlikely to generate any evidence of safety or effectiveness. Once a product is granted EUA and administered to patients, it is unlikely that any useful information will be obtained to benefit future patients, because there is no systematic data collection on effectiveness or safety.
Based on all this very clear information from the CDC/FDA and the IMNA, it would be fair to conclude that Emergency Use Authorization is a process that should be applied very judiciously and only in cases of dire emergencies.
Now let’s look at what types of emergency situations EUA is legally designed to address.
EAU is meant for WMD emergencies
The laws permitting the EUA “Access Mechanism” described above were drawn up for cases of extreme, immediate emergencies involving weapons of mass destruction (WMD), also referred to as CBRN (chemical, biological, radiological, nuclear) agents.
Here’s how the Food & Drug Administration (FDA) describes its EUA powers:
Section 564 of the FD&C Act (21 U.S.C. 360bbb–3) allows FDA to strengthen public health protections against biological, chemical, nuclear, and radiological agents.
With this EUA authority, FDA can help ensure that medical countermeasures may be used in emergencies to diagnose, treat, or prevent serious or life-threatening diseases or conditions caused by biological, chemical, nuclear, or radiological agents when there are no adequate, approved, and available alternatives (among other criteria).
These EUA powers were granted in 2004 under very specific circumstances related to preparedness for attacks by CBRN agents.
As explained in Harvard Law’s Bill of Health,
Ultimately, it was the War on Terror that would give rise to emergency use authorization. After the events of September 11, 2001 and subsequent anthrax mail attacks, Congress enacted the Project Bioshield Act of 2004.
The record indicates that Congress was focused on the threat of bioterror specifically, not on preparing for a naturally-occurring pandemic.
Given such a narrow type of truly extreme emergency situation involving a WMD attack, it is understandable why the EUA “access mechanism” does not require a lot of regulatory oversight or adherence to any manufacturing or clinical trial standards.
So what the EUA access mechanism actually require?
The 3 Steps for Emergency Use Authorization (EUA)
Three things have to happen in order for EUA to be granted to a medical product:
The Secretary of Homeland Security, the Secretary of Defense, or the Secretary of Health and Human Services needs to determine that there is an emergency involving an attack or a threat of an attack with a CBRN agent or a disease caused by such an agent.
The FDA needs to make sure that it meets four “statutory criteria” when it issues the EUA.
The FDA has to “impose certain required conditions” in the EUA.
EUA Step 1: Declaring a CBRN emergency
The emergency declaration for EUA is separate and unrelated to any other emergency declarations that may be issued by the President, the HHS Secretary, or anyone else. It must be issued specifically for the purpose of activating EUA and can be ended or extended independently of any other emergency declaration.
Here’s what the EUA law states are the four possible scenarios for activating the EUA “access mechanism”:
a determination by the Secretary of Homeland Security that there is a domestic emergency, or a significant potential for a domestic emergency, involving a heightened risk of attack with a biological, chemical, radiological, or nuclear agent or agents;
a determination by the Secretary of Defense that there is a military emergency, or a significant potential for a military emergency, involving a heightened risk to United States military forces, including personnel operating under the authority of Title 10 or Title 50, of attack with—
a biological, chemical, radiological, or nuclear agent or agents; or
an agent or agents that may cause, or are otherwise associated with, an imminently life-threatening and specific risk to United States military forces;
a determination by the Secretary [of Health and Human Services] that there is a public health emergency, or a significant potential for a public health emergency, that affects, or has a significant potential to affect, national security or the health and security of United States citizens living abroad, and that involves a biological, chemical, radiological, or nuclear agent or agents, or a disease or condition that may be attributable to such agent or agents; or
the identification of a material threat pursuant to section 319F–2 of the Public Health Service Act [42 U.S.C. 247d–6b] sufficient to affect national security or the health and security of United States citizens living abroad.
EUA Step 2. Meeting the statutory criteria
Once one of the secretaries has declared that there is an emergency that warrants EUA, there are four more “statutory criteria” that have to be met in order for the FDA to issue the EUA. Here’s how the FDA explains these requirements:
Serious or Life-Threatening Disease or Condition
For FDA to issue an EUA, the CBRN agent(s) referred to in the HHS Secretary’s EUA declaration must be capable of causing a serious or life-threatening disease or condition.
Evidence of Effectiveness
Medical products that may be considered for an EUA are those that "may be effective" to prevent, diagnose, or treat serious or life-threatening diseases or conditions that can be caused by a CBRN agent(s) identified in the HHS Secretary’s declaration of emergency or threat of emergency under section 564(b).
The "may be effective" standard for EUAs provides for a lower level of evidence than the "effectiveness" standard that FDA uses for product approvals. FDA intends to assess the potential effectiveness of a possible EUA product on a case-by-case basis using a risk-benefit analysis, as explained below.
[BOLDFACE ADDED]
Risk-Benefit Analysis
A product may be considered for an EUA if the Commissioner determines that the known and potential benefits of the product, when used to diagnose, prevent, or treat the identified disease or condition, outweigh the known and potential risks of the product.
In determining whether the known and potential benefits of the product outweigh the known and potential risks, FDA intends to look at the totality of the scientific evidence to make an overall risk-benefit determination. Such evidence, which could arise from a variety of sources, may include (but is not limited to): results of domestic and foreign clinical trials, in vivo efficacy data from animal models, and in vitro data, available for FDA consideration. FDA will also assess the quality and quantity of the available evidence, given the current state of scientific knowledge.
[BOLDFACE ADDED]
No Alternatives
For FDA to issue an EUA, there must be no adequate, approved, and available alternative to the candidate product for diagnosing, preventing, or treating the disease or condition. A potential alternative product may be considered “unavailable” if there are insufficient supplies of the approved alternative to fully meet the emergency need.
EUA Step 3. Imposing the required conditions
Once we have the EUA-specific emergency declaration, and once the FDA determines that the product may be effective and that whatever evidence is available shows that its benefits outweigh its risks, there is one more layer of related regulation.
Here’s how a 2018 Congressional Research Service report on EUA explains this:
FFDCA §564 directs FDA to impose certain required conditions in an EUA and allows for additional discretionary conditions where appropriate. The required conditions vary depending upon whether the EUA is for an unapproved product or for an unapproved use of an approved product. For an unapproved product, the conditions of use must:
(1) ensure that health care professionals administering the product receive required information;
(2) ensure that individuals to whom the product is administered receive required information;
(3) provide for the monitoring and reporting of adverse events associated with the product; and
(4) provide for recordkeeping and reporting by the manufacturer.
CONCLUSION
As noted in this article, the FDA/CDC clearly recognize that the process of granting Emergency Use Authorization (EUA) is unlikely to generate any information about the effectiveness or safety of a product. When we look at the letter of the law governing EUA, we see that this is, indeed, a correct assessment.
The EUA law does not impose any legal or regulatory standards that might determine whether a product is safe or effective. The only standards are whether the FDA believes the product may be effective and that its known benefits outweigh its known harms. If there are no known harms or known benefits, because the product has never been through the drug approval process, the FDA can use whatever information or standards it chooses to make that determination.
It follows from all of this that a company whose product is a candidate for EUA may attempt to demonstrate the product’s safety and/or effectiveness through whatever means it chooses. The existence of such an attempt (whether a clinical trial or other data-collecting mechanism), and how that attempt is conducted, are all up to the company. Nothing in the EUA law applies to how the company designs, conducts or analyzes any studies or other data-collecting mechanisms it chooses to pursue.
Applied to Covid products this means:
No safety or efficacy data from clinical trials were required in order for Covid products to receive EUA.
Any clinical trials referred to in the EUA process were conducted with no legally applicable regulatory standards.
When we find out that these products lack efficacy or safety, that is not a surprise. It is a highly likely result of the process.
There is no data from the EUA process on which to base non-EUA decisions about safety or efficacy of the product. So any non-EUA use of the product would require going through the legal approval process for regular medical products from the beginning.
More on the approval process for Covid vaccines here.
"No informed consent is required, but patients who “volunteer” to take the product must be told they can refuse and that the product is unapproved/available under EUA." This is most disturbing. Doesn't this rule automatically prohibit the use of vaccine mandates? What am I missing?
The EUA was a fraud.. there were alternative JUST not the ones that Big Pharma wanted